Mutations
Beta-thalassemia
Beta-thalassemia is a result of deletions in the HBB gene. This deletion can encompass only the β-globin gene or it can include the whole β-globin gene cluster (OMIM - Beta-Thalassemia, MIM# 613985, 2011). Beta-zero-thalassemia results when there are no detectable β- globin chains present. When there are low detectable amounts of the β-globin chains available, the condition is called Beta-plus-thalassemia. The absence or low concentration of β-globin causes the subsequent decrease in the normal complete human Hemoglobin molecule (HbA) and hence anemia in the patient (OMIM - Beta-Thalassemia, MIM# 613985, 2011). The most common populations affected are people of the Middle East, Mediterranean, India, Far East, Central Asia and Transcaucasus (OMIM - Beta-Thalassemia, MIM# 613985, 2011).
Sickle Cell Anemia
Sickle cell anemia is a growing health concern because statistics have shown that approximately 7% of the population has the mutation and between 300,000 and 400,000 affected children are born each year (Rusanova et al., 2011). It is the most common disorder caused by a single nucleotide change (GAG → GTG) resulting in the sixth amino acid change from glutamic acid to valine (Rusanova et al., 2011; Kitayama Cabral et al., 2011; Ashley-Koch et al., 2000). This mutation results in a gene product called HbS which is a malformed protein (Ashley-Koch et al., 2000). The HbS protein subunits do not fold properly and form a long, rigid molecule which forces the red blood cells into a sickle shape (Fig 1). Primarily, these sickle shaped blood cells prematurely die and cause anemia. Affected blood cells also block small blood vessels and cause organ damage and pain (Ashley-Koch et al., 2000; US Library of Medicine, 2011; Nucleotide, NCBI- Human HBB mRNA, 2011; OMIM - Sickle Cell Anemia, MIM# 603903, 2011).
Sickle cell anemia is a growing health concern because statistics have shown that approximately 7% of the population has the mutation and between 300,000 and 400,000 affected children are born each year (Rusanova et al., 2011). It is the most common disorder caused by a single nucleotide change (GAG → GTG) resulting in the sixth amino acid change from glutamic acid to valine (Rusanova et al., 2011; Kitayama Cabral et al., 2011; Ashley-Koch et al., 2000). This mutation results in a gene product called HbS which is a malformed protein (Ashley-Koch et al., 2000). The HbS protein subunits do not fold properly and form a long, rigid molecule which forces the red blood cells into a sickle shape (Fig 1). Primarily, these sickle shaped blood cells prematurely die and cause anemia. Affected blood cells also block small blood vessels and cause organ damage and pain (Ashley-Koch et al., 2000; US Library of Medicine, 2011; Nucleotide, NCBI- Human HBB mRNA, 2011; OMIM - Sickle Cell Anemia, MIM# 603903, 2011).
Figure 1: Sickle cell and normal phenotype of the human red blood cell. (Gabriel, Abram M.D. & Jennifer Przybylski, 2010)
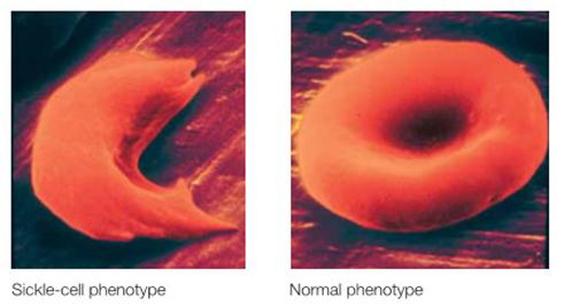
Individuals whom carry only one HbS allele have what is called sickle cell trait. In this state, individuals do not express sickle cell but are carriers of the disease. Individuals with two copies of the HbS gene are affected by the disease (Ashley-Koch et al., 2000; OMIM - Sickle Cell Anemia, MIM# 603903, 2011). Specifically, there are different haplotypes, or specific mutations recognized by restriction endonucleases in the beta gene cluster (Kitayama Cabral et al., 2011; Ashley-Koch et al., 2000; Cardoso and Guerreiro, 2010). The most commonly occurring haplotypes are named after the location of which they were discovered: Bantu (or Central African Republic (CAR)), Senegal, Saudi Arabia-India, Benin and Camaroon (Fig. 2) (Kitayama Cabral et al., 2011; Ashley-Koch et al., 2000; Gabriel, Abram M.D. & Jennifer Przybylski, 2010; Cardoso and Guerreiro, 2010).
Figure 2: Distribution of SCA haplotypes in Africa and surrounding areas. Individual pie charts for each area studied represent relative frequencies of the haplotypes. CAR = Central African Republic. (Gabriel, Abram M.D. & Jennifer Przybylski, 2010)
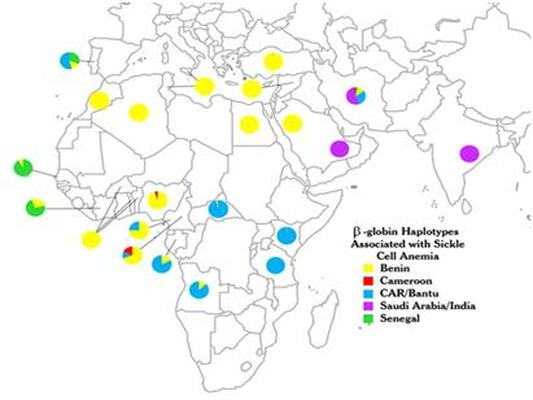
Haplotypes linked to the HbS allele assist in predicting sickle cell anemia complications in patients (Cardoso and Guerreiro, 2010). Patients with the Bantu (CAR) haplotype are present with the most severe clinical symptoms, whereas the Senegal and Arab-India haplotypes have the mildest symptoms. The Benin and Cameroon haplotypes are intermediate in severity (Kitayama Cabral et al., 2011; Cardoso and Guerreiro, 2010). The gradation regarding symptom severity is closely tied to the fetal hemoglobin levels in the blood, where the Senegal and Arab-India haplotypes produce the highest levels and the Bantu mutation presents with the lowest concentration in the blood (Kitayama Cabral et al., 2011). Fetal hemoglobin levels are thought to neutralize the negative effects of the mutated beta globin gene product (Gabriel, Abram M.D. & Jennifer Przybylski, 2010).
Medicines and Genetic Treatments
To combat sickle cell disease, medicines are being tested to increase the fetal hemoglobin levels of gamma globin molecules found in the beta globin gene cluster. Chemicals, such as hydroxyurea (aka hyroxycarbamide) and Azacytamide, have been shown to boost the levels of gamma globin and in turn also prevent polymerization of the HbS protein subunits and hence sickling of the blood cells (OMIM - Sickle Cell Anemia, MIM# 603903, 2011).
Genetic strategies have also been tested to treat the diseases associated with mutations and deletions of the human HBB gene. These methods include: 1) additions of an “HBB like gene” to haematopoietic stem cells to use for bone marrow transplantation, 2) reactivation of the endogenous HBG gene (gamma globin) using oligonucleotides instead of chemicals, 3) targeting the mutant beta globin transcript by strategies, such as RNA interference and 4) gene repair or addition at the HBB gene by homologous recombination (Mansilla-Soto et al, 2011).
The most promising strategy to be used in clinical trials has been shown to be the transduction of the HBB like gene into haematopoietic stem cells (HSC- stems cells that give rise to all types of blood cells) (Mansilla-Soto et al, 2011). However, these studies have demonstrated that there are possible side effects and risks. Insertional oncogenesis has occurred with the use of retroviral vectors which can result in the development of leukemia. The patient may also reject the HSC transplantation. Further studies need to be completed to make transduction of the HBB like gene safer by using lentiviral vectors, instead of retroviruses. This vector would provide a more specific gene insertion and less risks resulting in oncogenesis (Mansilla-Soto et al, 2011). Furthermore, studies using pluripotent stems cells may be a more effective area of study, providing they will be approved for clinical use (Mansilla-Soto et al, 2011).
Medicines and Genetic Treatments
To combat sickle cell disease, medicines are being tested to increase the fetal hemoglobin levels of gamma globin molecules found in the beta globin gene cluster. Chemicals, such as hydroxyurea (aka hyroxycarbamide) and Azacytamide, have been shown to boost the levels of gamma globin and in turn also prevent polymerization of the HbS protein subunits and hence sickling of the blood cells (OMIM - Sickle Cell Anemia, MIM# 603903, 2011).
Genetic strategies have also been tested to treat the diseases associated with mutations and deletions of the human HBB gene. These methods include: 1) additions of an “HBB like gene” to haematopoietic stem cells to use for bone marrow transplantation, 2) reactivation of the endogenous HBG gene (gamma globin) using oligonucleotides instead of chemicals, 3) targeting the mutant beta globin transcript by strategies, such as RNA interference and 4) gene repair or addition at the HBB gene by homologous recombination (Mansilla-Soto et al, 2011).
The most promising strategy to be used in clinical trials has been shown to be the transduction of the HBB like gene into haematopoietic stem cells (HSC- stems cells that give rise to all types of blood cells) (Mansilla-Soto et al, 2011). However, these studies have demonstrated that there are possible side effects and risks. Insertional oncogenesis has occurred with the use of retroviral vectors which can result in the development of leukemia. The patient may also reject the HSC transplantation. Further studies need to be completed to make transduction of the HBB like gene safer by using lentiviral vectors, instead of retroviruses. This vector would provide a more specific gene insertion and less risks resulting in oncogenesis (Mansilla-Soto et al, 2011). Furthermore, studies using pluripotent stems cells may be a more effective area of study, providing they will be approved for clinical use (Mansilla-Soto et al, 2011).
